| 出品公司: | Agilent |
|---|---|
| 载体名称: | pVPack-Ampho |
| 质粒类型: | 哺乳动物载体;逆病毒包装载体;双质粒包装系统;信封载体 |
| 高拷贝/低拷贝: | 高拷贝 |
| 克隆方法: | 限制性内切酶,多克隆位点 |
| 启动子: | CMV |
| 载体大小: | 6.8kb |
| 5' 测序引物及序列: | CMV fwd 5’CGCAAATGGGCGGTAGGCGTG 3’ |
| 3' 测序引物及序列: | -- |
| 载体标签: | 无 |
| 载体抗性: | Ampicillin |
| 筛选标记: | 嘌呤霉素 |
| 克隆菌株: | DH5α或 HB101 |
| 宿主细胞(系): | 包装细胞系如293T |
| 备注: |
逆病毒包装载体pVPack-Ampho是2质粒包装系统的信封质粒, 与pVPack-GP一起使用,适用范围、使用方法见下文。 |
| 产品目录号: | #217568 |
| 稳定性: | 稳表达 |
| 组成型/诱导型: | 组成型 |
| 病毒/非病毒: | 非病毒 |
pVPack-Ampho
价格:元
规格: 2ug质粒(仅用于转化提取) 转染级去内毒素液体即用质粒100μg 转染级去内毒素液体即用质粒500μg 转染级去内毒素液体即用质粒1mg 更大规格-质粒构建-质粒定制-请联系我们
联系方式:I47-825O-882O
买家导航

pVPack-Ampho载体质粒基本信息
pVPack-Ampho质粒图谱载体图谱和pVPack-Ampho载体序列质粒序列多克隆位点信息
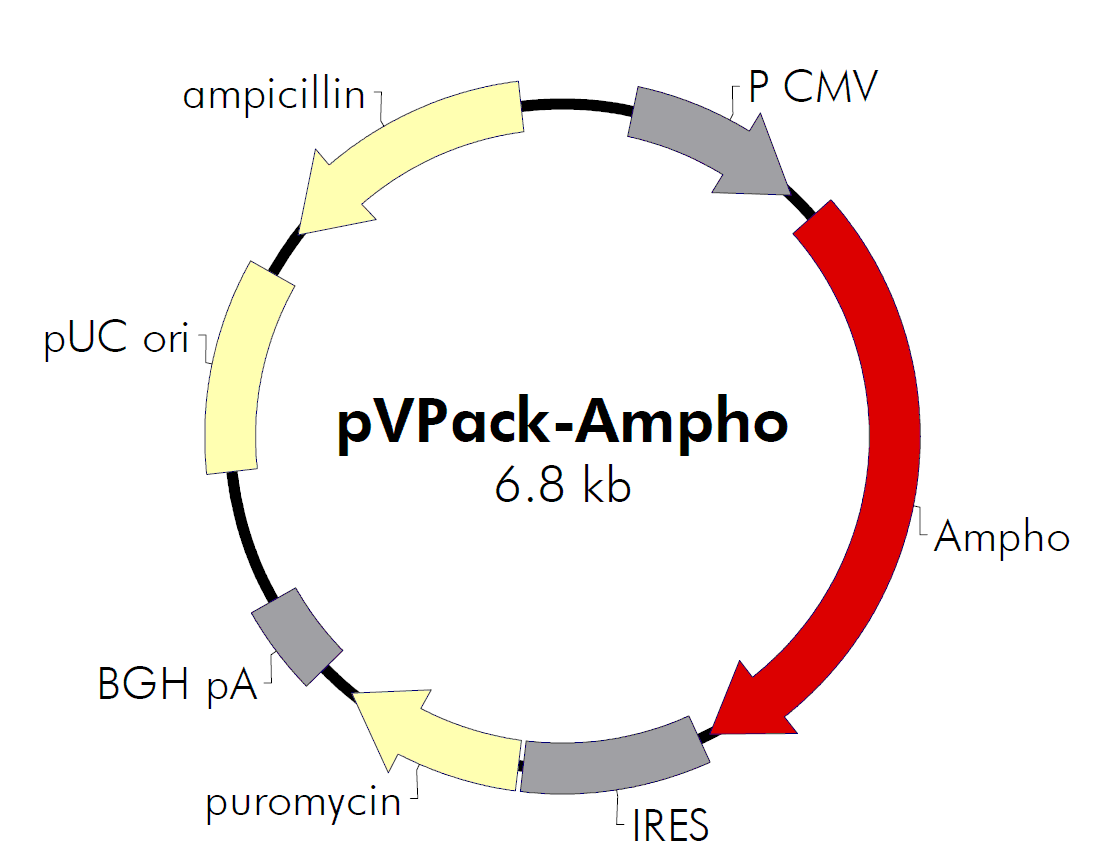
pVPack-Ampho质粒载体简介
pVPack逆病毒包装载体描述
Choice of env-Expressing Vector
In addition to the gag-pol expression vector pVPack-GP, the pVPack vector system offers 4 different env-expressing vectors. Which of those 4 is selected depends on the choice of host cell type; see Table I and Miller (1997).2 The pVPack-Eco vector is the safest vector, providing experiments can be performed in transduced mouse or rat cells; ecotropic virus infects human cells with extremely low efficiency. The amphotropic envelope protein has historically been the protein of choice for infection of human and other mammalian cell lines. More recently the 10A1 envelope protein has been used due to its increased versatility relative to the amphotropic protein. The 10A1 protein recognizes the same cell-surface receptor as the amphotropic envelope protein plus a second receptor, and thus can essentially infect any cell that an amphotropic virus can infect, although in some cases with a higher efficiency. The ecotropic, amphotropic and 10A1 proteins are all natural MMLV variants, and are all relatively labile and thus considered relatively safe compared with other viral systems. The vesicular stomatitis virus G protein (VSV-G) is rapidly becoming the most popular envelope protein. Unlike the other three MMLV-derived envelope proteins which recognize cell surface receptors, VSV-G recognizes a phospholipid that is present on all cell types, and thus can theoretically allow the efficient infection of any mitotic cell.3 Special precautions must be used when working with this vector (see Preprotocol Safety Considerations).
Vector Features
Figures 2 and 3 illustrates the important features of the vectors in the pVPack system. The expression of both the gag-pol elements in the pVPack-GP vector and the envelope elements in the env-expressing vectors are driven by the CMV promoter. Each of these vectors also contains an internal ribosome entry site (IRES) linked to a downstream drug-resistance cassette that enables the selection of stable producer lines. The vector pVPack-GP and the env-expressing vectors employ different resistance cassettes, hisD and puromycin, respectively. Methods for selecting stable producer lines are discussed in Hartman and Mulligan (1988)4 and Wirth and colleagues (1988).5
Notes
If a compatible MMLV-based retroviral vector is chosen, all three vectors can be maintained simultaneously. Although stable VSV-G-expressing cells lines have been successfully constructed, in general they are of poor quality due to the toxicity of VSV-G. For the production of VSV-G pseudotyped virus, transient transfection rather than selection of stable cell lines is recommended.
The bacterial origin of replication, pUC, and ampicillin resistance cassette are included to permit maintenance and production of the vectors in E. coli.
Choice of Expression Vector
Any MMLV-based retroviral vector for gene delivery and expression can be used with the pVPack vector system to produce high-titer retroviral stocks. The Stratagene pFB and pCFB retroviral vectors and ViraPort retroviral cDNA libraries are compatible with the Stratagene pVPack system. They contain the elements necessary for virion packaging: a bacterial origin of replication and ampicillin-resistance gene from pBR322, an extended MMLV packaging signal (ψ+), and a multiple cloning site (MCS) that is located between the MMLV 5′ and 3′ long terminal repeat sequences (LTRs).
pFB-Neo-LacZ, pFB-hrGFP, and pFB-Luc Control Vectors
The pFB-Neo-LacZ plasmid vector provided with the kit contains a bicistronic transcript; the β-galactosidase gene is expressed from the first open reading frame, and is followed by the neomycin-resistance marker downstream from an IRES. The vector may be used as an expression control, and can also be used to determine viral titer by FACS, in situ staining with X-gal, or G418-resistant colony formation.
Also available for use as a positive control is the vector pFB-hrGFP (Stratagene Catalog #240027), which contains coding sequence for the humanized green fluorescent protein from a novel marine organism. The hrGFP-expressing vector can be used to determine the transfection efficiency of the packaging cell line and to determine viral titer by FACS.
The pFB-Luc control* (also available separately) allows a qualitative assessment of the efficiency with which the target cell type is transduced by retrovirus. Direct comparisons between the cell lines based on luciferase activity should be made with caution however, as differences in luciferase activity may be due to cell type-dependent differences in luciferase expression rather than differences in transduction efficiencies.

双质粒系统逆病毒包装载体pVPack-Ampho使用方法
PROTOCOL FOR RETROVIRUS PRODUCTION
Prior to production of virus, users should be thoroughly familiar with the suggestions and Web sites described in the section Preprotocol Safety Considerations. All virus work should be performed in a designated virus work area. All cell lines to be used for production of or infection by retrovirus should first be tested for the presence of endogenous retrovirus. See Undesired Production of Replication-Competent Retrovirus above.
Although a variety of protocols and cell lines may be successfully used with these vectors, the following protocol for the production of viral supernatants is recommended. This protocol consistently results in the production of viral titers >107 colony forming units (cfu)/ml when transducing NIH3T3 cells with a pFB-derived vector. The protocol employs a calcium phosphate precipitation of the vector DNA and is based on the Transfection MBS Mammalian transfection kit, modified according to Pear and colleagues.7 Although excellent results may be obtained using 293 cells, we recommend the use of the 293 cell derivative 293T, which has been shown to transfect with a significantly greater efficiency.7
Day 1: Preparing for Production of Virus by Transfection
293T Host Cell Preparation
Split 293T cells at 2.5-3.0 × 106 cells per 60-mm tissue culture plate in growth medium (See Preparation of Media and Reagents) 24 hours before the transfection and incubate at 37°C until needed.
Note To achieve optimal titers, it is important that the 293T cells are healthy and growing exponentially. Cells should be passaged at high density, and ideally passaged no more than 20 times (no more than approximately 2 months); it is thus prudent to initially prepare a large number of frozen vials of the cells while they are at a low passage and healthy. Care should be taken to avoid clumping of the cells during passaging and plating for transfection.
Plasmid DNA Preparation
DNA preparations of high purity should be used for the transfections.
1. Pipette the following into a clean 1.5-ml microcentrifuge tube; prepare one tube for each transfection to be carried out. ♦3 μg an MMLV-based retroviral plasmid containing the gene of interest ♦3 μg pVPack-GP (gag-pol-expressing vector) ♦3 μg of one of the four env-expressing vectors (pVPack-Eco, pVPack-Ampho, pVPack-VSV-G, pVPack-10A1)
2. (Optional) Prepare the positive control vector sample by pipetting the following into a clean 1.5-ml microcentrifuge tube.
♦3 μg pFB-Neo-LacZ or pFB-hrGFP ♦3 μg pVPack-GP (gag-pol-expressing vector) ♦3 μg of one of the four env-expressing vectors (pVPack-Eco, pVPack-Ampho, pVPack-VSV-G, pVPack-10A1)
3. To each of the tubes containing the mixed vector DNA, add 1 ml 100% (v/v) ethanol and 0.1 × volume 3 M sodium acetate to the DNA mixture; mix by inverting the tube, incubate at -80°C for 30 minutes. Collect the DNA pellet by centrifugation at 12,000 × g for 10 minutes at 4°C. Aspirate and discard the supernatant. Add 1 ml 70% (v/v) ethanol to the tube, vortex briefly, and collect the DNA pellet by centrifugation at 12,000 × g for 5 minutes at 4°C. Remove and discard the supernatant; close the cap of the tube. Store wet pellets at 4°C overnight.
Day 2: Transfecting Cells
Note The procedure on Day 2 will take a minimum of 10 hours to complete.
Adding the MBS-Containing Medium to the Cells
1 Inspect the host cells that were split the day before; they should be approximately 80% confluent. [If cells are significantly less than 80% confluent, viral supernatants may be harvested 72 hours post-transfection rather than 48 hours.]
2 Prepare the MBS-containing medium (see Preparation of Media and Reagents). This must be done immediately prior to the transfection. For each 60-mm tissue culture plate, 4 ml of MBS-containing medium must be prepared.
3 Add 4 ml of MBS-containing medium to each 60-mm plate and return the plates to the 37°C incubator. This must be done 20-30 minutes before the addition of the DNA suspension.
Adding the DNA Suspension to the Cells
1 Remove the microcentrifuge tubes containing the wet DNA pellets (including the pFB-Neo-LacZ or pFB-hrGFP–containing pellet if the control transfection is to be carried out) from storage at 4°C and transfer them to the laminar flow hood.
2 Resuspend each DNA pellet in 450 μl sterile H2O and transfer the liquid to separate 5-ml BD Falcon polystyrene round-bottom tubes.
3 To each resuspended DNA pellet add 50 μl of Solution I and 500 μl Solution II from the Transfection MBS Mammalian Transfection Kit.
4 Gently resuspend any precipitate in the DNA suspension by pipetting the suspension up and down with a pipettor set at 500 μl. The DNA suspension should appear clear to opaque. Allow the DNA suspension to sit at room temperature for 10 minutes.
5 Remove the 60-mm plates to be transfected from the incubator and add the DNA suspension onto the plates in a dropwise fashion, swirling gently to prevent the cells from being lifted from the plate and to distribute the DNA suspension evenly.
Note From this point on, it should be assumed that infectious virus is present in the supernatant of the transfected cells. Gloves and disposable lab coats should be worn while working with the virus. We recommend that gloved hands be sprayed intermittently with ethanol. When pipetting medium supernatant and transferring plates to and from the laminar flow hood, aerosols should be avoided. All dirty pipets and plasticware should be disposed of as described in the section
6 Return the tissue culture plates to the 37°C incubator.
7 After incubating for 3 hours, remove the medium from the plates and replace it with 4 ml of growth medium supplemented with 25 μM chloroquine. Return the plates to the 37°C incubator.
8 After incubating for an additional 6–7 hours, remove the growth medium containing 25 μM chloroquine and replace with 4 ml growth medium—no chloroquine.
Day 3: Preparing for the Transduction
1 Remove growth medium from 293T plates and replace with 3.0 ml of fresh growth medium. Return the plates to the 37°C incubator.
Note If virus is to be harvested 72 hours post-transfection rather than 48 hours, steps 2 and 3 should be carried out on Day 4.
2 Split the target cells, seeding 1 × 105 cells per well for 6-well plates and 2 × 104 cells per well for 24-well plates. This seeding density may vary with the cell line; 20–30% confluency is desirable.
3 Return the plates to the 37°C incubator overnight.
Day 4: Transducing the Target Cells
Note If virus is to be harvested 72 hours post-transfection rather than 48 hours, all steps from the Day 4 section should be performed on Day 5.
1. Remove the virus-producing 293T cells from the incubator.
2. Collect the virus-containing supernatant from the first plate and filter it through a 0.45 μm filter into a sterile 50-ml conical tube.
Note If desired, the supernatant can be snap frozen on dry ice or liquid nitrogen and stored at -80°C at this stage. WARNING: Freeze-thawing virus one time typically results in a 2-fold loss in titer. Subsequent freeze-thaw cycles result in less than a 2-fold loss per cycle of the remaining infectious virus.
3 Dilute viral supernatants as desired in growth medium.
4 Add DEAE-dextran solution to the diluted viral supernatants to a final concentration of 10 μg/ml (1:1000 dilution of the 10 mg/ml DEAE-dextran stock.
Note If starting from a frozen supernatant stock, thaw rapidly in a 37°C water bath, minimizing the time the supernatant is at 37°C before the addition of the DEAE-dextran.
5 Remove the plates containing the target cells from the incubator.
6 Remove and discard the medium from the wells of the target cell plates.
a) Add DEAE-dextran plus virus to the wells containing the target cells:
b) 1.0 ml per well for 6-well plates and 200 μl per well for 24-well plates.
7 Return the plates to the 37°C incubator for 3 hours.
8 After the 3 hour incubation, add growth medium to the wells: 1.0 ml per well for 6-well plates and 200 μl per well for 24-well plates.
Note
For expression studies, allow at least two days between target cell infection and cell harvest.
pVPack-Ampho质粒序列载体序列
pVPack-Ampho载体图谱质粒图谱pdf版和pVPack-Ampho载体序列质粒序列等相关资料下载
pVPack-Ampho质粒载体应用举例
- 上一篇:pVPack-10A1
- 下一篇:pMSCV-PIG

